UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
CURRENT REPORT
Pursuant to Section 13 or 15(d)
of the Securities Exchange Act of 1934
Date of report (Date of earliest event reported):
(Exact Name of Registrant as Specified in its Charter)
| (State or Other Jurisdiction of Incorporation) |
(Commission File Number) |
(IRS Employer Identification No.) |
| |
||
| (Address of Principal Executive Offices) | (Zip Code) |
Registrant’s telephone number, including area code:
Not applicable
(Former Name or Former Address, if Changed Since Last Report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions:
| Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class |
Trading |
Name of each exchange on which registered | ||
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§240.12b-2 of this chapter).
Emerging growth company
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act.
| Item 2.02. | Results of Operations and Financial Condition. |
On August 14, 2025, Invivyd, Inc. (the “Company”) issued a press release announcing its financial results for the quarter ended June 30, 2025, and recent business highlights. A copy of the press release is furnished as Exhibit 99.1 to this Current Report on Form 8-K and is incorporated by reference into this Item 2.02.
The information furnished pursuant to this Item 2.02, including Exhibit 99.1, shall not be deemed to be “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liabilities of that Section, nor shall it be deemed to be incorporated by reference into any of the Company’s filings with the Securities and Exchange Commission under the Exchange Act or the Securities Act of 1933, as amended, whether made before or after the date hereof, regardless of any general incorporation language in such a filing, except as expressly set forth by specific reference in such a filing.
| Item 8.01. | Other Events. |
On August 14, 2025, the Company issued a press release entitled “Invivyd Aligns with U.S. FDA on Rapid Pathway to Full Approval (BLA) of Vaccine Alternative Monoclonal Antibody VYD2311 to Protect American Adults and Adolescents from COVID-19.” A copy of the press release is filed herewith as Exhibit 99.2 and is incorporated by reference into this Item 8.01.
On August 14, 2025, the Company posted an updated corporate presentation on its website at www.invivyd.com. A copy of the presentation is filed as Exhibit 99.3 to this Current Report on Form 8-K and is incorporated by reference into this Item 8.01.
| Item 9.01. | Financial Statements and Exhibits. |
(d) Exhibits
| Exhibit No. |
Description | |
| 99.1 | Press Release, dated August 14, 2025 | |
| 99.2 | Press Release, dated August 14, 2025 | |
| 99.3 | Corporate Presentation, dated August 14, 2025 | |
| 104 | Cover Page Interactive Data File (embedded within the Inline XBRL document) | |
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| INVIVYD, INC. | ||||||
| Date: August 14, 2025 | By: | /s/ Jill Andersen | ||||
| Jill Andersen | ||||||
| Chief Legal Officer and Corporate Secretary | ||||||
Exhibit 99.1

Invivyd Reports Second Quarter 2025 Financial Results and Recent Business Highlights
| • | PEMGARDA® (pemivibart) net product revenue of $11.8 million reported for Q2 2025, representing 413% growth year-over-year |
| • | Invivyd’s target of near-term profitability (1H 2025) was not met but remains possible with the upcoming respiratory virus season |
| • | Announced alignment with U.S. FDA on rapid pathway to full approval (BLA) of vaccine alternative monoclonal antibody candidate VYD2311 to protect American adults and adolescents from COVID-19 |
| • | Announced attractive safety profile and pharmacokinetics data for VYD2311 from our pre-pivotal first-in-human clinical trial, including 76-day observed half-life for IM route of administration, allowing for potential long-term protection |
WALTHAM, Mass., August 14, 2025 – Invivyd, Inc. (Nasdaq: IVVD) today announced financial results for the quarter ended June 30, 2025, and provided recent business highlights.
“We believe we are entering a remarkable period of change for the company that points toward an exciting future,” said Marc Elia, Chairman of the Board at Invivyd. “Having characterized VYD2311 safety and pharmacokinetics in our Phase 1/2 trial, we are fully engaged on designing our clinical and go-to-market strategy for what we believe will be an exciting alternative to COVID-19 vaccination. As PEMGARDA® (pemivibart) moves into the fall season, we expect to move quickly to cement our position as the leading provider of monoclonal antibody technology for Americans in need of COVID-19 protection. With U.S. Food and Drug Administration (FDA) advice in hand, we look forward to the VYD2311 Phase 2/3 clinical trial design and finalization with the FDA, including anticipated alignment on a pivotal study plan for pediatrics, and advancement of other pipeline opportunities that leverage our technology, such as RSV and measles.”
“While the second quarter PEMGARDA growth was modest, the base business is growing along with our internal commercial capabilities, while we are simultaneously looking toward an improved target product profile with VYD2311 to protect ordinary Americans from symptomatic COVID-19,” said Bill Duke, Chief Financial Officer of Invivyd. “In addition to further protocol development to advance VYD2311, we look forward to additional upcoming anticipated milestones including identification of a potentially best-in-class RSV candidate in Q3 2025, identification of a preclinical measles candidate by the end of 2025, and advancement of our efforts to support the Long COVID community with the recently formed SPEAR Study Group.”
Recent Business Highlights
| • | Commercial Execution |
| • | PEMGARDA® (pemivibart) uptake continues to grow among healthcare providers caring for immunocompromised patients, supported by Invivyd’s in-house sales force and expanded field presence across key specialties, even during period of low COVID-19 transmission. |
| • | Pemivibart has been added to the National Comprehensive Cancer Network® (NCCN®) Clinical Practice Guidelines in Oncology for B-Cell Lymphomas. |
| • | Clinical & Regulatory Developments |
| • | In June 2025, Invivyd announced positive full Phase 1/2 clinical data for VYD2311, a next-generation monoclonal antibody (mAb) candidate designed to prevent and treat COVID-19, including an attractive safety profile demonstrated across all dosing cohorts and routes of administration – intravenous (IV), subcutaneous (SC), and intramuscular (IM); all reported adverse events (AEs) were deemed unrelated or classified as mild to moderate and largely related to injection site and infusion reactions with no serious or severe AEs observed. |
| • | Following a single dose, serum concentrations of VYD2311 remained high at six months with an observed half-life of the IM dose route having the longest duration at 76.0 (CI: 68.5 – 90.7) days. |
| • | Comprehensive dose modeling of VYD2311 serum virus neutralizing antibody (sVNA) titers (submitted to FDA and recently published in medRxiv) indicates possible strong protection from symptomatic COVID-19 achievable via IM dosing on a long interval (months to quarters and beyond). |
| • | Following a Type C meeting in July 2025, Invivyd announced alignment with FDA on a rapid pathway to full approval (BLA) of vaccine alternative monoclonal antibody VYD2311 to protect American adults and adolescents from COVID-19, subject to agreement on safety database size for Phase 2/3 clinical trial and pending full protocol review, and announced the company’s objectives for VYD2311 development. |
| • | Biologics License Application (BLA) pathway for VYD2311 to be supported by a single, Phase 2/3 randomized, double-blind, placebo-controlled trial with a primary endpoint of reduction in symptomatic COVID-19, resembling CANOPY Cohort B. |
| • | Company’s target product profile for VYD2311 is low-dose, IM, scalable, low-cost, long-lasting, protective option for target populations, including adults and adolescents (12 years+; 40kg+) and, subject to FDA alignment, pediatrics (aged 0 to 12 years). |
| • | Company anticipates compact trial (12-week primary endpoint analysis) evaluating prevention of COVID-19 largely among ordinary Americans, enabling rapid enrollment. |
| • | Planned head-to-head safety evaluation of VYD2311 with COVID-19 vaccine, pending regulatory alignment. |
| • | Pipeline Expansion |
| • | Company has initiated discovery efforts to assess pipeline expansion beyond SARS-CoV-2, including potential targets such as respiratory syncytial virus (RSV) and measles. |
| • | Company anticipates providing an update on identification of an RSV candidate in the third quarter of 2025 and an update on identification of a preclinical measles mAb candidate in the fourth quarter of 2025. |
| • | Corporate and Financial Updates |
| • | In July 2025, Invivyd and leading researchers formed SPEAR (Spike Protein Elimination and Recovery) Study Group to assess the effects of mAb therapy for Long COVID and COVID-19 Post-Vaccination Syndrome. |
| • | In April 2025, Invivyd entered into a $30 million non-dilutive term loan facility with Silicon Valley Bank, a division of First Citizens Bank, supporting balance sheet optionality and providing potential additional runway for commercial and pipeline execution if certain conditions and milestones are met. |
Second Quarter 2025 Financial Results:
| • | Revenue: Reported Q2 2025 PEMGARDA net product revenue of $11.8 million, as compared to $2.3 million in Q2 2024. |
| • | Cash Position: Cash and cash equivalents were $34.9 million as of June 30, 2025. |
| • | Research & Development (R&D) Expenses (including In-Process R&D): R&D expenses were $9.6 million for the quarter ended June 30, 2025, compared to $30.3 million for the comparable period of 2024. This decrease is primarily attributable to a decrease in commercial manufacturing costs of VYD2311, a decrease in clinical trial costs related to our CANOPY Phase 3 clinical trial and a decrease in personnel-related costs. |
| • | Selling, General & Administrative (SG&A) Expenses: SG&A expenses were $16.6 million for the quarter ended June 30, 2025, compared to $21.1 million for the comparable period of 2024. This decrease is primarily attributable to a decrease in stock-based compensation expense, partially offset by increased headcount-related costs and professional services fees. |
| • | Net Loss and Net Loss per Share: Net loss was $14.7 million for the quarter ended June 30, 2025, compared to $47.2 million for the comparable period in 2024. Basic and diluted net loss per share was $0.12 for the quarter ended June 30, 2025, compared to $0.40 for the comparable period in 2024. |
About PEMGARDA
PEMGARDA® (pemivibart) is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and provided evidence of clinical efficacy in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19. PEMGARDA has demonstrated in vitro neutralizing activity against major SARS-CoV-2 variants, including JN.1, KP.3.1.1, XEC and LP.8.1. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb that has not been approved, but has been authorized for emergency use by the U.S. FDA under an EUA for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
PEMGARDA is not authorized for use for treatment of COVID-19, treatment of Long COVID, or post-exposure prophylaxis of COVID-19. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination.
Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse reactions included systemic infusion-related reactions and hypersensitivity reactions, local infusion site reactions, and infusion site infiltration or extravasation. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including important safety information and boxed warning.
To support the EUA for PEMGARDA, an immunobridging approach was used to determine if PEMGARDA may be effective for pre-exposure prophylaxis of COVID-19. Immunobridging is based on the serum virus neutralizing titer-efficacy relationships identified with other neutralizing human mAbs against SARS-CoV-2. This includes adintrevimab, the parent mAb of pemivibart, and other mAbs that were previously authorized for EUA. There are limitations of the data supporting the benefits of PEMGARDA. Evidence of clinical efficacy for other neutralizing human mAbs against SARS-CoV-2 was based on different populations and SARS-CoV-2 variants that are no longer circulating. Further, the variability associated with cell-based EC50 value determinations, along with limitations related to pharmacokinetic data and efficacy estimates for the mAbs in prior clinical trials, impact the ability to precisely estimate protective titer ranges. Additionally, certain SARS-CoV-2 viral variants may emerge that have substantially reduced susceptibility to PEMGARDA, and PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants.
The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies.
About CANOPY
The CANOPY Phase 3 clinical trial was designed to evaluate the safety and tolerability of pemivibart and to assess immunobridging from pemivibart to certain historical data from the company’s previous Phase 2/3 clinical trial of adintrevimab (ADG20) for the prevention of symptomatic COVID-19 (EVADE). Additionally, there were pre-specified exploratory endpoints through three, six and twelve months to evaluate clinical efficacy of pemivibart compared to placebo in the prevention of RT-PCR-confirmed symptomatic COVID-19. The latest analysis from the Phase 3 CANOPY clinical trial included 365-day data. The CANOPY clinical trial enrolled participants in two cohorts: Cohort A was a single-arm, open-label trial in adults with moderate-to-severe immune compromise including complex underlying medical conditions. Cohort B was a randomized, placebo-controlled cohort that enrolled adults without moderate-to-severe immune compromise at risk of acquiring COVID-19 due to regular unmasked face-to-face interactions in indoor settings.
About VYD2311
VYD2311 is a novel monoclonal antibody (mAb) candidate being developed for COVID-19 to continue to address the urgent need for new prophylactic and therapeutic options. The pharmacokinetic profile and antiviral potency of VYD2311 may offer the ability to deliver clinically meaningful titer levels through more patient-friendly means such as an intramuscular route of administration.
VYD2311 was engineered using Invivyd’s proprietary integrated technology platform and is the product of serial molecular evolution designed to generate an antibody optimized for neutralizing contemporary virus lineages. VYD2311 leverages the same antibody backbone as pemivibart, Invivyd’s investigational mAb granted emergency use authorization in the U.S. for the pre-exposure prophylaxis (PrEP) of symptomatic COVID-19 in certain immunocompromised patients, and adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and demonstrated clinically meaningful results in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19.
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2. Invivyd deploys a proprietary integrated technology platform unique in the industry designed to assess, monitor, develop, and adapt to create best in class antibodies. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for a monoclonal antibody (mAb) in its pipeline of innovative antibody candidates. Visit https://invivyd.com/ to learn more.
Trademarks are the property of their respective owners.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipates,” “believes,” “could,” “expects,” “estimates,” “intends,” “potential,” “predicts,” “projects,” and “future” or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, the company’s target of achieving profitability; expectations regarding the COVID-19 landscape and upcoming respiratory virus season; beliefs about the company’s market position; plans related to the company’s research and development activities; expectations regarding the biophysical properties, clinical trial design, regulatory pathway, target product profile and target populations for VYD2311; the anticipated focus and goals of the SPEAR Study Group; the ongoing in vitro neutralizing activity of PEMGARDA against dominant SARS-CoV-2 variants; the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in certain immunocompromised persons; the potential of VYD2311 as a novel mAb candidate that may be able to deliver clinically meaningful titer levels through more patient-friendly means; the company’s devotion to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2; potential pipeline expansion beyond SARS-CoV-2, including potential targets such as RSV and measles, and expected announcements related thereto; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: uncertainties regarding the company’s expectations, projections and estimates regarding future costs and expenses, future revenue, capital requirements, and the availability of and the need for additional financing; whether the company’s cash and cash equivalents are sufficient to support its operating plan for as long as anticipated; uncertainties regarding market acceptance, payor coverage and reimbursement, or future revenue generated by PEMGARDA; how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether such EUA is revised or revoked by the FDA; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; the success of the company’s in-house sales force, and company’s ability to maintain and expand sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; changes in expected or existing competition; changes in the regulatory environment; the outcome of the company’s engagement with regulators; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways; uncertainties related to reaching agreement with FDA on the safety database size for the pivotal clinical trial of VYD2311 and full protocol review; clinical trial site activation or enrollment rates; the timing, progress and results of the company’s discovery, preclinical and clinical development activities; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; variability of results in models and methods used to predict activity against SARS-CoV-2 variants; whether the epitope that pemivibart and VYD2311 targets remains structurally intact; whether the company’s product candidates are able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; whether the company’s integrated technology platform is able to produce mAbs with broad and durable viral protection along with improved drug properties; the company’s reliance on third parties; complexities of manufacturing mAb therapies, and availability of quantities of commercial product in the future, if authorized or approved; macroeconomic and political uncertainties; the company’s ability to realize the anticipated benefits of its term loan facility; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this press release are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2024 and the company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2025, each as filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations
(781) 208-1747
media@invivyd.com
Investor Relations
(781) 208-1747
investors@invivyd.com
INVIVYD, INC.
CONDENSED CONSOLIDATED BALANCE SHEETS
(UNAUDITED)
(In thousands, except share and per share amounts)
| June 30, 2025 |
December 31, 2024 |
|||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 34,905 | $ | 69,349 | ||||
| Accounts receivable, net |
8,698 | 10,906 | ||||||
| Prepaid expenses and other current assets |
15,678 | 20,426 | ||||||
|
|
|
|
|
|||||
| Total current assets |
59,281 | 100,681 | ||||||
| Inventory |
25,440 | 25,907 | ||||||
| Property and equipment, net |
1,392 | 1,508 | ||||||
| Operating lease right-of-use assets |
3,010 | 1,385 | ||||||
| Other non-current assets |
15 | 34 | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 89,138 | $ | 129,515 | ||||
|
|
|
|
|
|||||
| Liabilities, Preferred Stock and Stockholders’ Equity |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 18,051 | $ | 10,448 | ||||
| Accrued expenses (1) |
25,307 | 50,197 | ||||||
| Operating lease liabilities |
1,067 | 1,304 | ||||||
| Other current liability |
26 | 27 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
44,451 | 61,976 | ||||||
|
|
|
|
|
|||||
| Operating lease liabilities, non-current |
1,898 | — | ||||||
|
|
|
|
|
|||||
| Total liabilities |
46,349 | 61,976 | ||||||
|
|
|
|
|
|||||
| Commitments and contingencies |
||||||||
| Stockholders’ equity: |
||||||||
| Preferred stock (undesignated), $0.0001 par value; 10,000,000 shares authorized and no shares issued and outstanding at June 30, 2025 and December 31, 2024 |
— | — | ||||||
| Common stock, $0.0001 par value; 1,000,000,000 shares authorized, 120,142,811 shares issued and outstanding at June 30, 2025; 119,835,162 shares issued and outstanding at December 31, 2024 |
12 | 12 | ||||||
| Additional paid-in capital |
975,759 | 969,526 | ||||||
| Accumulated other comprehensive loss |
(39 | ) | (5 | ) | ||||
| Accumulated deficit |
(932,943 | ) | (901,994 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ equity |
42,789 | 67,539 | ||||||
|
|
|
|
|
|||||
| Total liabilities, preferred stock and stockholders’ equity |
$ | 89,138 | $ | 129,515 | ||||
|
|
|
|
|
|||||
| (1) | Includes related-party amounts of $490 and $1,274 as of June 30, 2025 and December 31, 2024, respectively. |
INVIVYD, INC.
CONDENSED CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(UNAUDITED)
(In thousands, except share and per share amounts)
| Three Months Ended June 30, |
Three Months Ended June 30, |
Six Months Ended June 30, |
Six Months Ended June 30, |
|||||||||||||
| 2025 | 2024 | 2025 | 2024 | |||||||||||||
| Revenue: |
||||||||||||||||
| Product revenue, net |
$ | 11,786 | $ | 2,264 | $ | 23,090 | $ | 2,264 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total revenue |
11,786 | 2,264 | 23,090 | 2,264 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Operating costs and expenses: |
||||||||||||||||
| Cost of product revenue (1) |
685 | 88 | 1,519 | 88 | ||||||||||||
| Research and development (2) |
9,573 | 30,334 | 20,214 | 61,494 | ||||||||||||
| Selling, general and administrative |
16,588 | 21,089 | 33,339 | 36,018 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total operating costs and expenses |
26,846 | 51,511 | 55,072 | 97,600 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Loss from operations |
(15,060 | ) | (49,247 | ) | (31,982 | ) | (95,336 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Other income: |
||||||||||||||||
| Other income, net |
400 | 2,000 | 1,033 | 4,593 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Total other income, net |
400 | 2,000 | 1,033 | 4,593 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss |
(14,660 | ) | (47,247 | ) | (30,949 | ) | (90,743 | ) | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Other comprehensive income (loss) |
||||||||||||||||
| Unrealized (loss) gain, net of tax |
(26 | ) | — | (34 | ) | 1 | ||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Comprehensive loss |
$ | (14,686 | ) | $ | (47,247 | ) | $ | (30,983 | ) | $ | (90,742 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Net loss per share attributable to common stockholders, basic and diluted |
$ | (0.12 | ) | $ | (0.40 | ) | $ | (0.26 | ) | $ | (0.77 | ) | ||||
|
|
|
|
|
|
|
|
|
|||||||||
| Weighted-average common shares outstanding, basic and diluted |
120,016,132 | 119,362,670 | 119,950,172 | 117,490,439 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| (1) | Includes related-party amounts of $472 and $924 for the three and six months ended June 30, 2025 and no related-party amounts for the three and six months ended June 30, 2024. |
| (2) | Includes related-party amounts of $1,140 and $2,268 for the three and six months ended June 30, 2025 and $1,131 and $2,266 for the three and six months ended June 30, 2024, respectively. |
Exhibit 99.2
Invivyd Aligns with U.S. FDA on Rapid Pathway to Full Approval (BLA) of Vaccine Alternative Monoclonal Antibody VYD2311 to Protect American Adults and Adolescents from COVID-19
| • | Alignment follows Type C meeting for VYD2311 as previously disclosed |
| • | BLA pathway for VYD2311 to be supported by a single, Phase 2/3 randomized, double-blind, placebo-controlled trial with a primary endpoint of reduction in symptomatic COVID-19, resembling CANOPY Cohort B |
| • | Company’s target product profile is low-dose, intramuscular, scalable, low-cost, long-lasting, protective option for target populations, including adults and adolescents (12 years+; 40kg+) and, subject to FDA alignment, pediatrics (aged 0 to 12 years) |
| • | Anticipate compact trial (12-week primary endpoint analysis) evaluating prevention of COVID-19 largely among ordinary Americans, enabling rapid enrollment |
| • | Planned head-to-head safety evaluation with COVID-19 vaccine, pending regulatory alignment |
| • | Quantities of VYD2311 potential commercial launch product available |
Waltham, Mass., August 14, 2025 – Invivyd, Inc. (Nasdaq: IVVD) today announced it has received and is aligned with advice from the U.S. Food and Drug Administration (FDA) on a compact and, therefore, rapid pathway to potential Biologics License Application (BLA) approval for Invivyd’s novel monoclonal antibody (mAb) candidate VYD2311, for the prevention of COVID-19. As part of a recent Type C meeting, FDA advised that a single, Phase 2/3 randomized, double-blind, placebo-controlled trial evaluating mAb efficacy from a relatively modest number of RT-PCR -confirmed symptomatic COVID-19 disease events could support a BLA submission for VYD2311 for the prevention of COVID-19 in a broad population of Americans (12 years of age and older, weighing at least 40kg), including immunocompromised people, subject to agreement on safety database size and pending full protocol review.
“We believe monoclonal antibodies such as VYD2311 can serve as a powerful alternative to vaccines for COVID-19 prevention, and represent an important potential paradigm shift to move American medicine beyond the real and perceived limitations of COVID-19 vaccines. Amidst declining public trust in vaccines, we want to offer Americans a new, non-vaccine choice,” said Marc Elia, Chairman of Invivyd’s Board of Directors. “COVID-19 monoclonal antibody medicines work alongside natural human immunity without needing to activate the immune system, and, if we can make them widely available, we believe mAbs represent the natural next step beyond vaccination to keep people safe and well, if approved, in the face of pervasive COVID-19.”
FDA advice and background statements to Invivyd cited observations on Invivyd’s efficient execution of its Phase 3 randomized clinical trial of pemivibart for the prevention of COVID-19 (CANOPY), the antiviral durability of pemivibart, and the strong protection demonstrated by pemivibart among ordinary Americans in CANOPY Cohort B, while recognizing that Invivyd antibodies stem from a common molecular lineage. Multiple such Invivyd antibodies have undergone successful randomized, placebo-controlled clinical trials, including efficacy assessment in contemporary Americans. In agreement with FDA advice, Invivyd plans to study two doses of VYD2311 to assess any differences in resulting levels of protection or differences in safety, with a goal to unlock further choice for Americans in need of COVID-19 protection.
“We are grateful for the FDA’s clear and constructive feedback, which provides a well-defined path forward for our COVID-19 development program. We believe the FDA’s feedback underscores the shared urgency to advance innovative solutions for prevention of COVID-19,” said Rachael Gerlach, Ph.D., Vice President, Regulatory Affairs at Invivyd. “Combined with Invivyd’s unique discovery platform, this alignment is a critical step in bringing forward a potentially important, medically attractive and patient-friendly alternative to COVID vaccination, providing protective monoclonal antibodies, if approved, to any American who wishes protection, whether immune compromised, at high risk for severe disease, or just interested in not getting sick.”
The FDA provided advice for pursuit of a traditional BLA pathway for the prevention of COVID-19 caused by SARS-CoV-2 in adults and adolescents weighing at least 40 kg, recommending a Phase 2/3 randomized, double-blind, placebo-controlled trial with a primary endpoint of RT-PCR-confirmed symptomatic COVID-19, with a timepoint for measuring the primary endpoint coinciding with the expected duration of protection, anticipated as 12 weeks (3 months), and the potential for selection of an additional, longer duration timepoint, which Invivyd anticipates as 24 weeks (6 months).
Invivyd’s analysis of CANOPY clinical trial data and Cox Proportional Hazards modeling, combined with biophysical properties of VYD2311, suggest likely robust, long-term protection from symptomatic COVID-19 due to high potency and observed long half-life of ~76 days, following a relatively low dose of VYD2311 via intramuscular route of administration. Invivyd’s most recent analysis aligns well with multiple similar analyses spanning the majority of SARS-CoV-2 virus variation since emerging as a global human disease in 2020, lending confidence to dose selection and anticipated clinical benefit. In addition, as COVID-19 monoclonal antibodies represent additional immune support and do not require engagement of the human immune system with associated inflammation symptoms (reactogenicity), Invivyd anticipates, pending regulatory alignment, a clinical trial cohort exploring randomized, active-controlled safety and tolerability of VYD2311 compared to vaccination.
“Our completed first-in-human study of VYD2311 demonstrated high SARS-CoV-2 antiviral titers and an attractive safety profile at very high doses, well beyond doses we contemplate going forward with in development. We look forward to opening a U.S. IND and moving as quickly as possible to finalize a pivotal clinical trial design with the FDA,” said Mark Wingertzahn, Ph.D., Senior Vice President, Clinical Development at Invivyd, “If successful, we have the potential to change practice and provide consumers and central health authorities with an attractive alternative to COVID-19 vaccination. We anticipate sharing our plans as soon as possible once we finalize them with FDA.”
Invivyd has quantities of VYD2311 clinical supply and potential commercial launch product available.
About VYD2311
VYD2311 is a novel monoclonal antibody (mAb) candidate being developed for COVID-19 to continue to address the urgent need for new prophylactic and therapeutic options. The pharmacokinetic profile and antiviral potency of VYD2311 may offer the ability to deliver clinically meaningful titer levels through more patient-friendly means such as an intramuscular route of administration.
VYD2311 was engineered using Invivyd’s proprietary integrated technology platform and is the product of serial molecular evolution designed to generate an antibody optimized for neutralizing contemporary virus lineages. VYD2311 leverages the same antibody backbone as pemivibart, Invivyd’s investigational mAb granted emergency use authorization in the U.S. for the pre-exposure prophylaxis (PrEP) of symptomatic COVID-19 in certain immunocompromised patients, and adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and demonstrated clinically meaningful results in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19.
About PEMGARDA
PEMGARDA® (pemivibart) is a half-life extended investigational monoclonal antibody (mAb). PEMGARDA was engineered from adintrevimab, Invivyd’s investigational mAb that has a robust safety data package and provided evidence of clinical efficacy in global Phase 2/3 clinical trials for the prevention and treatment of COVID-19. PEMGARDA has demonstrated in vitro neutralizing activity against major SARS-CoV-2 variants, including JN.1, KP.3.1.1, XEC and LP.8.1. PEMGARDA targets the SARS-CoV-2 spike protein receptor binding domain (RBD), thereby inhibiting virus attachment to the human ACE2 receptor on host cells.
PEMGARDA (pemivibart) injection (4500 mg), for intravenous use is an investigational mAb that has not been approved, but has been authorized for emergency use by the U.S. FDA under an EUA for the pre-exposure prophylaxis (prevention) of COVID-19 in adults and adolescents (12 years of age and older weighing at least 40 kg) who have moderate-to-severe immune compromise due to certain medical conditions or receipt of certain immunosuppressive medications or treatments and are unlikely to mount an adequate immune response to COVID-19 vaccination. Recipients should not be currently infected with or have had a known recent exposure to an individual infected with SARS-CoV-2.
PEMGARDA is not authorized for use for treatment of COVID-19, treatment of Long COVID, or post-exposure prophylaxis of COVID-19. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination.
Anaphylaxis has been observed with PEMGARDA and the PEMGARDA Fact Sheet for Healthcare Providers includes a boxed warning for anaphylaxis. The most common adverse reactions included systemic infusion-related reactions and hypersensitivity reactions, local infusion site reactions, and infusion site infiltration or extravasation. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including important safety information and boxed warning.
To support the EUA for PEMGARDA, an immunobridging approach was used to determine if PEMGARDA may be effective for pre-exposure prophylaxis of COVID-19. Immunobridging is based on the serum virus neutralizing titer-efficacy relationships identified with other neutralizing human mAbs against SARS-CoV-2. This includes adintrevimab, the parent mAb of pemivibart, and other mAbs that were previously authorized for EUA. There are limitations of the data supporting the benefits of PEMGARDA. Evidence of clinical efficacy for other neutralizing human mAbs against SARS-CoV-2 was based on different populations and SARS-CoV-2 variants that are no longer circulating. Further, the variability associated with cell-based EC50 value determinations, along with limitations related to pharmacokinetic data and efficacy estimates for the mAbs in prior clinical trials, impact the ability to precisely estimate protective titer ranges. Additionally, certain SARS-CoV-2 viral variants may emerge that have substantially reduced susceptibility to PEMGARDA, and PEMGARDA may not be effective at preventing COVID-19 caused by these SARS-CoV-2 viral variants.
The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies.
About Invivyd
Invivyd, Inc. (Nasdaq: IVVD) is a biopharmaceutical company devoted to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2. Invivyd deploys a proprietary integrated technology platform unique in the industry designed to assess, monitor, develop, and adapt to create best in class antibodies. In March 2024, Invivyd received emergency use authorization (EUA) from the U.S. FDA for a monoclonal antibody (mAb) in its pipeline of innovative antibody candidates. Visit https://invivyd.com/ to learn more.
Trademarks are the property of their respective owners.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipates,” “believes,” “could,” “expects,” “estimates,” “intends,” “potential,” “predicts,” “projects,” and “future” or similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. Forward-looking statements include statements concerning, among other things, plans related to the company’s research and development activities; expectations regarding the biophysical properties, clinical trial design and enrollment, regulatory pathway, target product profile and target populations for VYD2311; the potential of VYD2311 as a novel mAb candidate that may be able to deliver clinically meaningful titer levels through more patient-friendly means; the expected advantages of mAbs such as VYD2311 as an alternative to COVID-19 vaccines, and Invivyd’s goal to provide Americans with choice; the company’s regulatory plans and the timing thereof, including expectations related to Invivyd’s engagement with regulators; anticipated future announcements about the VYD2311 program, and the timing thereof; the potential of PEMGARDA as a mAb for pre-exposure prophylaxis (prevention) of COVID-19 in certain immunocompromised persons; the company’s devotion to delivering protection from serious viral infectious diseases, beginning with SARS-CoV-2; and other statements that are not historical fact. The company may not actually achieve the plans, intentions or expectations disclosed in the company’s forward-looking statements and you should not place undue reliance on the company’s forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause the company’s actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: the timing, progress and results of the company’s discovery, preclinical and clinical development activities, including finalization of a pivotal clinical trial design for VYD2311 and initiation thereof; uncertainties related to agreement with FDA on the safety database size for the pivotal clinical trial of VYD2311 and full protocol review; clinical trial site activation or enrollment rates; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; variability of results in models and methods used to predict activity against SARS-CoV-2 variants; whether the epitope that VYD2311 and pemivibart targets remains structurally intact; whether the company’s product candidates are able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; changes in the regulatory environment; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways; the company’s ability to generate the clinical data needed to support a potential BLA submission for VYD2311; how long the EUA granted by the FDA for PEMGARDA will remain in effect and whether the EUA is revised or revoked by the FDA; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; changes in expected or existing competition; the company’s reliance on third parties; complexities of manufacturing mAb therapies, and availability of quantities of commercial launch product in the future, if authorized or approved; macroeconomic and political uncertainties; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this press release are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2024 and the company’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2025, each as filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward-looking statements contained in this press release are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law.
This press release contains hyperlinks to information that is not deemed to be incorporated by reference in this press release.
Contacts:
Media Relations
(781) 208-1747
media@invivyd.com
Investor Relations
(781) 208-1747
investors@invivyd.com

Exhibit 99.3 Corporate Deck August 2025 © 2025 Invivyd, Inc. Invivyd®, the Invivyd logo, Pemgarda® and the Ribbon logo are registered trademarks of Invivyd, Inc. All trademarks in this presentation are the property of their respective owners. 1

CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS This presentation contains forward-looking statements within the meaning of the U.S. Private Securities Litigation Reform Act of 1995. Statements in this presentation that are not statements of historical fact are forward-looking statements. Words such as “may,” “will,” “should,” “expect,” “plan,” “anticipate,” “seek,” “could,” “intend,” “target,” “aim,” “project,” “designed to,” “estimate,” “believe,” “predict,” “potential” or “continue” or the negative of these terms or other similar expressions are intended to identify forward-looking statements, though not all forward-looking statements contain these identifying words. Forward-looking statements include statements concerning, among other things, expectations about the COVID-19 landscape and impact on Americans; the belief that COVID-19 vaccines do not provide sufficient protection; the potential of monoclonal antibodies (mAbs) to robustly protect people from getting sick with COVID-19; the potential of PEMGARDA® (pemivibart) as a mAb for pre-exposure prophylaxis (PrEP) of COVID-19 in certain adults and adolescents with moderate-to-severe immune compromise; our plans, strategy and expectations related to the commercialization of PEMGARDA; the potential of VYD2311 as a high potency, long half life mAb with improved biophysical properties; our goals with respect to the target product profile of VYD2311 and expected advantages of VYD2311; estimates regarding the size of target patient populations and the potential market opportunity for our product candidates, as well as our market position; our research and clinical development efforts, including statements regarding initiation or completion of studies or trials, the time-frame during which results may become available, and the potential utility of generated data; our expectations regarding regulatory alignment with the U.S. Food and Drug Administration (FDA); the design of our proprietary platform to address evolution and broad medical need; the potential of our pipeline; our goals with respect to VYD25XY series mAbs and additional diseases and indications (e.g., RSV, measles, lyme); our business strategies and objectives, and ability to execute on them; our future prospects; future investments and anticipated R&D developments; our goal of near-term profitability; and other statements that are not historical fact. We may not actually achieve the plans, intentions or expectations disclosed in our forward-looking statements and you should not place undue reliance on our forward-looking statements. These forward-looking statements involve risks and uncertainties that could cause our actual results to differ materially from the results described in or implied by the forward-looking statements, including, without limitation: how long the emergency use authorization (EUA) granted by the FDA for PEMGARDA for COVID-19 PrEP in certain immunocompromised patients will remain in effect and whether such EUA is revised or revoked by the FDA; the ability to maintain a continued acceptable safety, tolerability and efficacy profile of any product candidate following regulatory authorization or approval; the success of the company’s in-house sales force, and company’s ability to maintain and expand sales, marketing and distribution capabilities to successfully commercialize PEMGARDA; changes in expected or existing competition; changes in the regulatory environment; the outcome of the company’s engagement with regulators; uncertainties related to the regulatory authorization or approval process, and available development and regulatory pathways; uncertainties related to reaching agreement with FDA on the safety database size for the pivotal clinical trial of VYD2311 and full protocol review; the timing, progress and results of the company’s discovery, preclinical and clinical development activities; unexpected safety or efficacy data observed during preclinical studies or clinical trials; the predictability of clinical success of the company’s product candidates based on neutralizing activity in nonclinical studies; the risk that results of nonclinical studies or clinical trials may not be predictive of future results, and interim data are subject to further analysis; the company’s reliance on third parties; potential variability in neutralizing activity of product candidates tested in different assays, such as pseudovirus assays and authentic assays; variability of results in models and methods used to predict activity against SARS-CoV-2 variants; whether the epitope that pemivibart and VYD2311 targets remains structurally intact; whether the company’s product candidates are able to demonstrate and sustain neutralizing activity against major SARS-CoV-2 variants, particularly in the face of viral evolution; whether the company’s integrated technology platform is able to produce mAbs with broad and durable viral protection along with improved drug properties; the complexities of manufacturing mAb therapies, and availability of quantities of commercial product in the future, if authorized or approved; macroeconomic and political uncertainties; uncertainties regarding the company’s expectations, projections and estimates regarding future costs and expenses, future revenue, capital requirements, and the availability of and the need for additional financing; whether the company’s cash and cash equivalents are sufficient to support its operating plan for as long as anticipated; uncertainties regarding market acceptance, payor coverage and reimbursement, or future revenue generated by PEMGARDA; the company’s ability to realize the anticipated benefits of its term loan facility; the company’s ability to continue as a going concern; and whether the company has adequate funding to meet future operating expenses and capital expenditure requirements. Other factors that may cause the company’s actual results to differ materially from those expressed or implied in the forward-looking statements in this presentation are described under the heading “Risk Factors” in the company’s Annual Report on Form 10-K for the year ended December 31, 2024, as filed with the Securities and Exchange Commission (SEC), and in the company’s other filings with the SEC, and in its future reports to be filed with the SEC and available at www.sec.gov. Forward-looking statements contained in this presentation are made as of this date, and Invivyd undertakes no duty to update such information whether as a result of new information, future events or otherwise, except as required under applicable law. 2

AGENDA u COVID-19 Landscape and Opportunity Next Steps 3
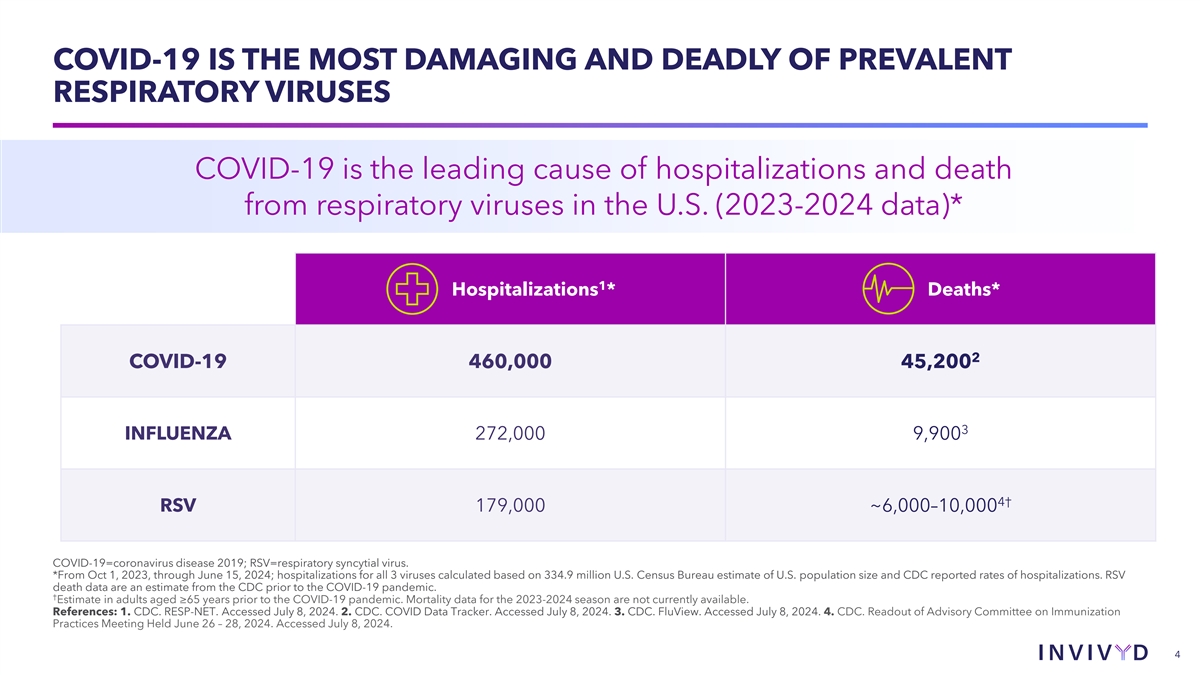
COVID-19 IS THE MOST DAMAGING AND DEADLY OF PREVALENT RESPIRATORY VIRUSES COVID-19 is the leading cause of hospitalizations and death from respiratory viruses in the U.S. (2023-2024 data)* 1 Hospitalizations * Deaths* 2 COVID-19 460,000 45,200 3 INFLUENZA 272,000 9,900 4† RSV 179,000 ~6,000–10,000 COVID-19=coronavirus disease 2019; RSV=respiratory syncytial virus. *From Oct 1, 2023, through June 15, 2024; hospitalizations for all 3 viruses calculated based on 334.9 million U.S. Census Bureau estimate of U.S. population size and CDC reported rates of hospitalizations. RSV death data are an estimate from the CDC prior to the COVID-19 pandemic. † Estimate in adults aged ≥65 years prior to the COVID-19 pandemic. Mortality data for the 2023-2024 season are not currently available. References: 1. CDC. RESP-NET. Accessed July 8, 2024. 2. CDC. COVID Data Tracker. Accessed July 8, 2024. 3. CDC. FluView. Accessed July 8, 2024. 4. CDC. Readout of Advisory Committee on Immunization Practices Meeting Held June 26 – 28, 2024. Accessed July 8, 2024. 4

COVID-”19”: AN ONGOING HEALTH CRISIS STILL MAKING HEADLINES YEAR AFTER YEAR 2025 2021 2022–2023 2024 COVID-19 cases are rising in these states amid summer wave, CDC data shows January 10, 2024 July 21, 2025 July 2, 2021 August 31, 2022 June 11, 2021 Why are more than 300 people in the US still dying from COVID every week? June 19, 2024 May 24, 2025 December 22, 2023 July 2, 2021 COVID rising in California. How bad will this summer be? January 23, 2024 July 28, 2025 August 12, 2021 December 22, 2023 All trademarks and logos displayed are the property of their respective owners. Their use here is for identification purposes only and does not constitute endorsement or affiliation. 5
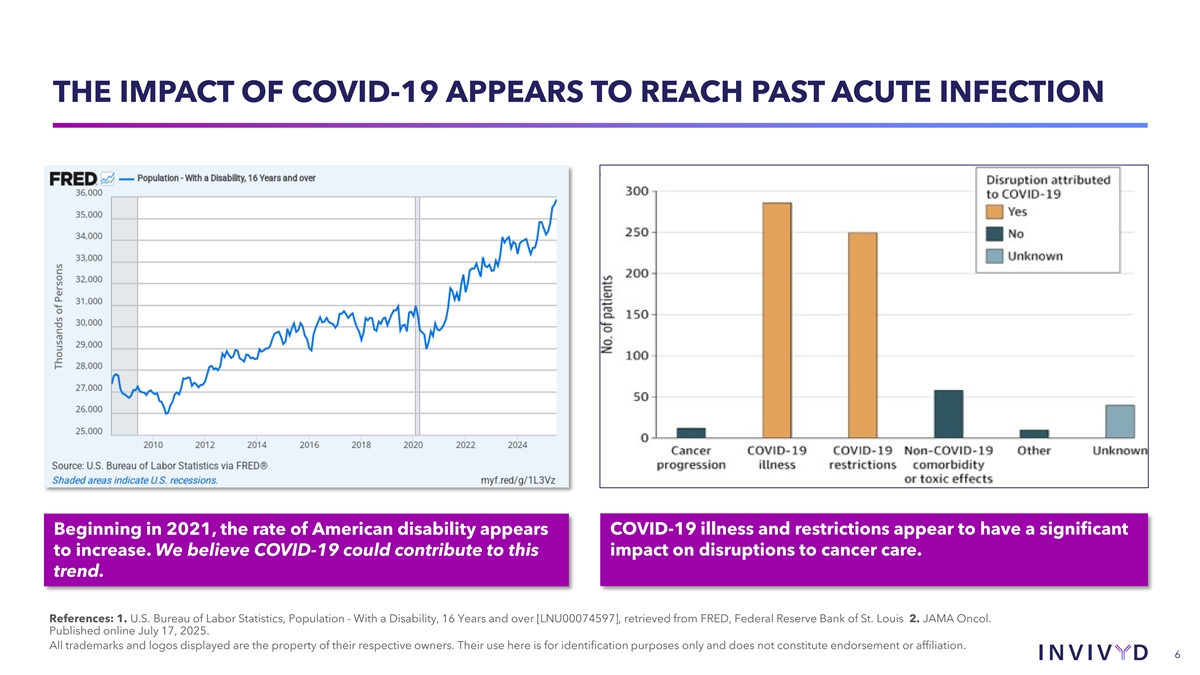
THE IMPACT OF COVID-19 APPEARS TO REACH PAST ACUTE INFECTION Beginning in 2021, the rate of American disability appears COVID-19 illness and restrictions appear to have a significant to increase. We believe COVID-19 could contribute to this impact on disruptions to cancer care. trend. References: 1. U.S. Bureau of Labor Statistics, Population - With a Disability, 16 Years and over [LNU00074597], retrieved from FRED, Federal Reserve Bank of St. Louis 2. JAMA Oncol. Published online July 17, 2025. All trademarks and logos displayed are the property of their respective owners. Their use here is for identification purposes only and does not constitute endorsement or affiliation. 6

AGENDA u COVID-19 u Landscape and Opportunity Next Steps 7
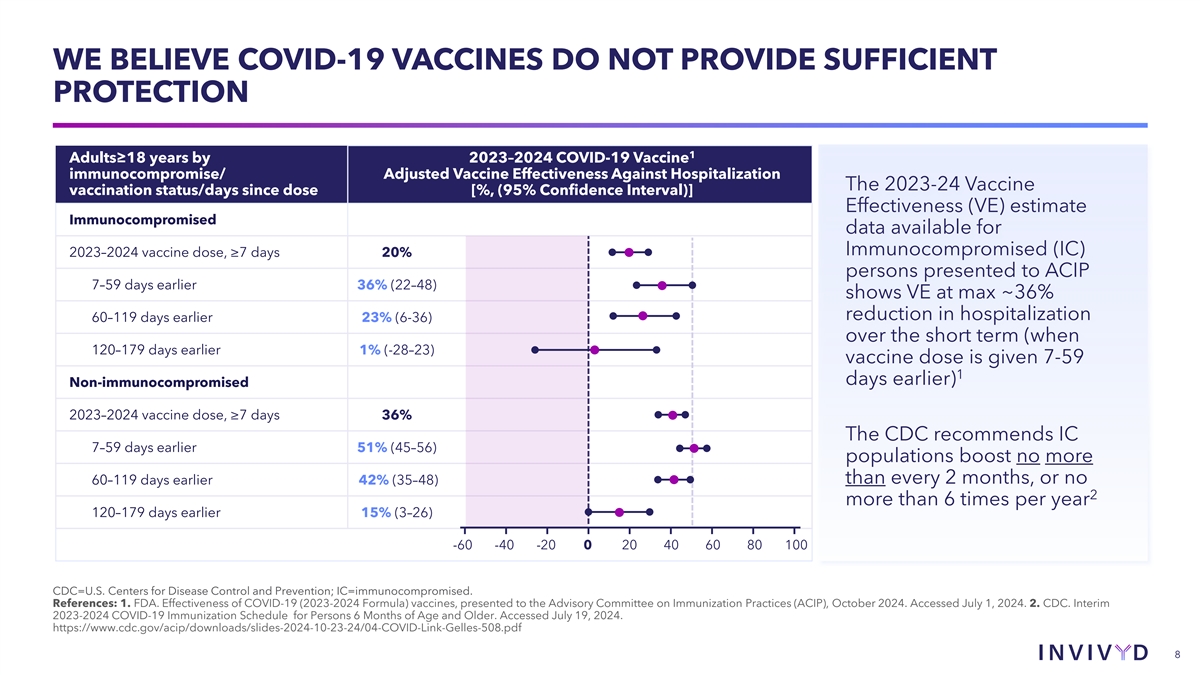
WE BELIEVE COVID-19 VACCINES DO NOT PROVIDE SUFFICIENT PROTECTION 1 Adults≥18 years by 2023–2024 COVID-19 Vaccine immunocompromise/ Adjusted Vaccine Effectiveness Against Hospitalization The 2023-24 Vaccine vaccination status/days since dose [%, (95% Confidence Interval)] Effectiveness (VE) estimate Immunocompromised data available for Immunocompromised (IC) 2023–2024 vaccine dose, ≥7 days 20% persons presented to ACIP 7–59 days earlier 36% (22–48) shows VE at max ~36% reduction in hospitalization 60–119 days earlier 23% (6-36) over the short term (when 120–179 days earlier 1% (-28–23) vaccine dose is given 7-59 1 days earlier) Non-immunocompromised 2023–2024 vaccine dose, ≥7 days 36% The CDC recommends IC 7–59 days earlier 51% (45–56) populations boost no more than every 2 months, or no 60–119 days earlier 42% (35–48) 2 more than 6 times per year 120–179 days earlier 15% (3–26) -60 -40 -20 0 20 40 60 80 100 CDC=U.S. Centers for Disease Control and Prevention; IC=immunocompromised. References: 1. FDA. Effectiveness of COVID-19 (2023-2024 Formula) vaccines, presented to the Advisory Committee on Immunization Practices (ACIP), October 2024. Accessed July 1, 2024. 2. CDC. Interim 2023-2024 COVID-19 Immunization Schedule for Persons 6 Months of Age and Older. Accessed July 19, 2024. https://www.cdc.gov/acip/downloads/slides-2024-10-23-24/04-COVID-Link-Gelles-508.pdf 8

MONOCLONAL ANTIBODIES HAVE BEEN SHOWN TO ROBUSTLY PROTECT PEOPLE FROM GETTING SICK WITH COVID-19 1 3,4 Tixagevimab+cilgavimab Adintrevimab * Pemivibart * 2 (“Evusheld”) * PROVENT 71% reduction* 84-94% reduction* 77% reduction* in risk of in risk of in risk of symptomatic COVID-19 symptomatic COVID-19 symptomatic COVID-19 through days 180 and 90, respectively, in ordinary Americans* *Figures provided represent relative risk reduction versus placebo in immunocompetent cohort References: 1. Ison MG, et al. Open Forum Infect Dis. 2023 Jun 13;10(7):ofad314. 2. Levin MJ, et al. N Engl J Med. 2022;386(23):2188-2200. 3. Invivyd. Data on File. 4. Symptomatic COVID-19 event collection in the CANOPY clinical trial is an exploratory endpoint and not part of the primary immunobridging endpoint of the CANOPY clinical trial. Adintrevimab is an investigational monoclonal antibody that has not been approved for use by any regulatory authorities; the safety and efficacy of adintrevimab have not been established. No head-to-head clinical trials have been conducted between adintrevimab, pemivibart, and/or tixagevimab+cilgavimab, and comparative conclusions cannot be made between antibodies. 9
OUR PROPRIETARY PLATFORM IS DESIGNED TO ADDRESS EVOLUTION AND BROAD MEDICAL NEED Rapid mAb Engineering Rapid, Compact Studies Commercial Opportunity • Multiple profiles anticipated to be generated for specific • Privileged epitopes • Compact clinical programs to molecules and use cases • Rapid affinity maturation evaluate mAbs expected to rapidly • We intend to consider diverse generate data for treatment and • Surveillance-informed metrics for routes of administration prevention use cases greater variation resistance (e.g., IV, IM, SC) for future mAb candidates • Goal to scale like vaccination IM=intramuscular; IV=intravenous; mAb=monoclonal antibody; SC=subcutaneous 10
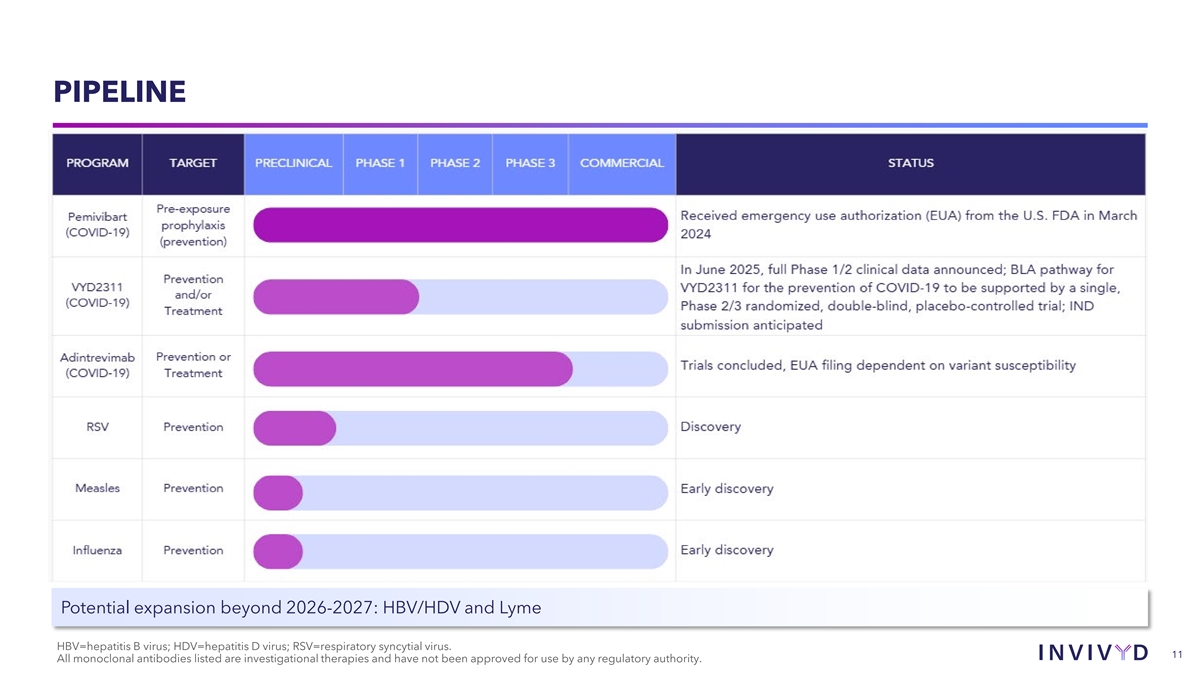
PIPELINE Potential expansion beyond 2026-2027: HBV/HDV and Lyme HBV=hepatitis B virus; HDV=hepatitis D virus; RSV=respiratory syncytial virus. 11 All monoclonal antibodies listed are investigational therapies and have not been approved for use by any regulatory authority.

PEMGARDA AUTHORIZED VIA EUA FOR THE PREVENTION OF COVID-19 FOR CERTAIN IMMUNOCOMPROMISED PERSONS PEMGARDA has not been approved but has been authorized for emergency use by the FDA under an emergency use authorization (EUA), for pre-exposure prophylaxis of COVID-19 in certain adults and adolescents (12 years of age and older weighing at least 40 kg) with moderate-to-severe immune compromise. Pre-exposure prophylaxis with PEMGARDA is not a substitute for vaccination in individuals for whom COVID-19 vaccination is recommended. Individuals for whom COVID-19 vaccination is recommended, including individuals with moderate-to-severe immune compromise who may derive benefit from COVID-19 vaccinations, should receive COVID-19 vaccination. In individuals who have recently received a COVID-19 vaccine, PEMGARDA should be administered at least 2 weeks after vaccination. The emergency use of PEMGARDA is only authorized for the duration of the declaration that circumstances exist justifying the authorization of the emergency use of drugs and biological products during the COVID-19 pandemic under Section 564(b)(1) of the Federal Food, Drug, and Cosmetic Act, 21 U.S.C. § 360bbb-3(b)(1), unless the declaration is terminated or authorization is revoked sooner. PEMGARDA is authorized for use only when the combined national frequency of variants with substantially reduced susceptibility to PEMGARDA is less than or equal to 90%, based on available information including variant susceptibility to PEMGARDA and national variant frequencies. For additional information, please see the PEMGARDA full product Fact Sheet for Healthcare Providers, including Important Safety Information and Boxed Warning. Reference: PEMGARDA [Fact Sheet for Healthcare Providers]. Invivyd; May 2025. 12 12

VYD2311 DESIGNED TO BE A HIGH POTENCY, LONG HALF LIFE MONOCLONAL ANTIBODY FOR COVID-19 Our next-generation mAb, VYD2311, designed to improve upon biophysical properties (potency, half-life) Development: Awaiting Pivotal Trial; low dose IM dosing possible • First-in-human (FIH) trial complete • Observed IM half-life of 76 days • Attractive safety profile demonstrated at high doses (well beyond target clinical dose), including: − 2g and 4.5g infusions (IV) − 1g intramuscular (IM) − 1.25g subcutaneous (SC) IM=intramuscular; IV=intravenous; SC = subcutaneous; mAb=monoclonal antibody Reference: Invivyd. Data on File. 13 13
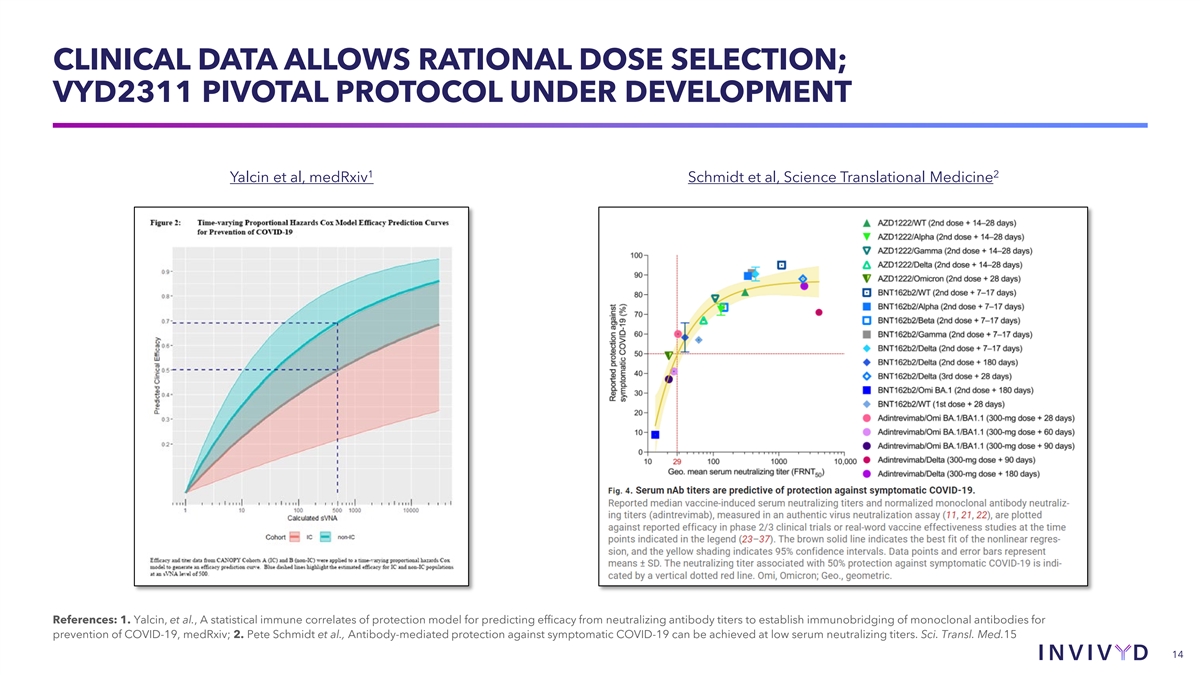
CLINICAL DATA ALLOWS RATIONAL DOSE SELECTION; VYD2311 PIVOTAL PROTOCOL UNDER DEVELOPMENT 1 2 Yalcin et al, medRxiv Schmidt et al, Science Translational Medicine References: 1. Yalcin, et al., A statistical immune correlates of protection model for predicting efficacy from neutralizing antibody titers to establish immunobridging of monoclonal antibodies for prevention of COVID-19, medRxiv; 2. Pete Schmidt et al., Antibody-mediated protection against symptomatic COVID-19 can be achieved at low serum neutralizing titers. Sci. Transl. Med.15 14 14

OUR GOAL: THE BEGINNING OF A NEW CATEGORY PEMGARDA VYD2311 Target Product Profile Status EUA BLA Population Mod / Severe IC Anyone Delivery High dose IV, multi-hour follow up Low dose IM (“boop”) at pharmacy / clinic Indication / Endpoint Immunobridge “Protection from COVID” Protection 84-94% Similar High dose IV with associated Safety IM, excellent hypersensitivity • COVID mAb: potential for equitable, • Planned head-to-head safety TIME TO GO instant, high, safe, natural, vaccine- demonstration to clearly distinguish free protection COVID mAb from COVID vaccine • Medical/social/political environment • PEMGARDA constraints removed for BIG potentially primed for disruption of VYD2311 = potential blockbuster COVID vaccine EUA=Emergency Use Authorization; BLA=Biologics License Application; IC=immunocompromised; IV=intravenous; IM=intramuscular; mAb=monoclonal antibody 15

GOAL: PROVIDE AMERICANS WITH A CHOICE FOR COVID-19 PROTECTION Monoclonal Antibody: COVID-19 Vaccine VYD2311 • Reactogenic • Not a vaccine • Myocarditis risk • Immunologically silent • Short duration of protection (weeks) • Natural, supplemental protection • Modest efficacy • Long duration of protection (months) • Was mandated • High efficacy (anticipated) • >35m doses administered in U.S. 2024 • Commercial launch quantities at-the-ready 16

INVIVYD IS ACTIVELY DRIVING TOWARD A SEISMIC SHIFT Rapid, efficient pathway unlocked to BLA submission for VYD2311 monoclonal antibody for COVID-19 prevention Invivyd makes minimally evolved molecular entity (“MEME”) monoclonal antibodies that are designed to minimize risk and have potential to enable efficient approval with a track record of predictable data from one molecule to the next (adintrevimab -> pemivibart -> VYD2311 -> VYD25xy …) Consistent data, consistent molecular structures, technical excellence, and better educating consumers on prevention = unlock of a new field of medicine: protective antibodies for whomever wants them, if approved 17

RESPIRATORY SYNCYTIAL VIRUS (RSV) DISCOVERY • Well-developed mAb medical category devoted to Fusion Protein Trimer prevention of RSV in neonates and children <24 months • Current key molecules: o Pavilizumab (1998) - AstraZeneca o Nirsevimab (2023) - Sanofi o Clesrovimab (2025 expected) - Merck • Opportunity to deploy Invivyd technology to create a best-in-class profile along one or more dimensions: o Neutralizing Potency (correlates to LRTI prevention rate) o Half-life o Barrier to resistance • Company anticipates providing an update on the Pantaleo Nat Rev Drug Dis 2022 identification of an RSV candidate in Q3 2025 LRTI = Lower Respiratory Tract Infection 18
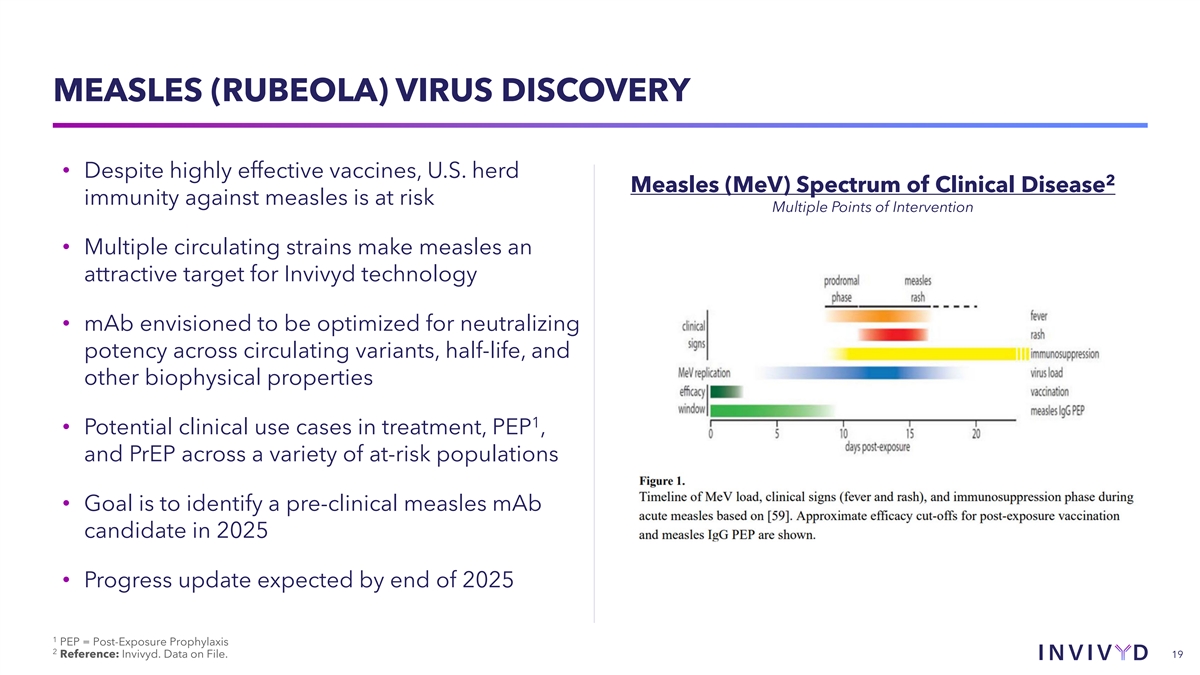
MEASLES (RUBEOLA) VIRUS DISCOVERY • Despite highly effective vaccines, U.S. herd 2 Measles (MeV) Spectrum of Clinical Disease immunity against measles is at risk Multiple Points of Intervention • Multiple circulating strains make measles an attractive target for Invivyd technology • mAb envisioned to be optimized for neutralizing potency across circulating variants, half-life, and other biophysical properties 1 • Potential clinical use cases in treatment, PEP , and PrEP across a variety of at-risk populations • Goal is to identify a pre-clinical measles mAb candidate in 2025 • Progress update expected by end of 2025 1 PEP = Post-Exposure Prophylaxis 2 Reference: Invivyd. Data on File. 19

AGENDA u COVID-19 u Landscape and Opportunity u Next Steps 20
AIMING FOR A DISRUPTION OF COVID-19 LANDSCAPE WHILE ADVANCING SELECT PIPELINE OPPORTUNITIES Design and align on pivotal clinical trial plan for PEMGARDA base business growing; informing pediatrics: potential for high-yield commercial commercial planning and supporting market focus (blockbuster Beyfortus® in pediatric RSV, preparation towards future portfolio growth but for COVID) Aiming for a disruption of COVID landscape Advance select pipeline opportunities 2026-2027 and beyond (2024: >35m vaccine balancing spend with cost of equity capital doses in U.S. = $3B+ revenue) Begin P2/3 VYD2311 clinical trial design and execution planning immediately for rapid finalization with global regulators All trademarks and logos displayed are the property of their respective owners. Their use here is for identification purposes only and does not constitute endorsement or affiliation. 21
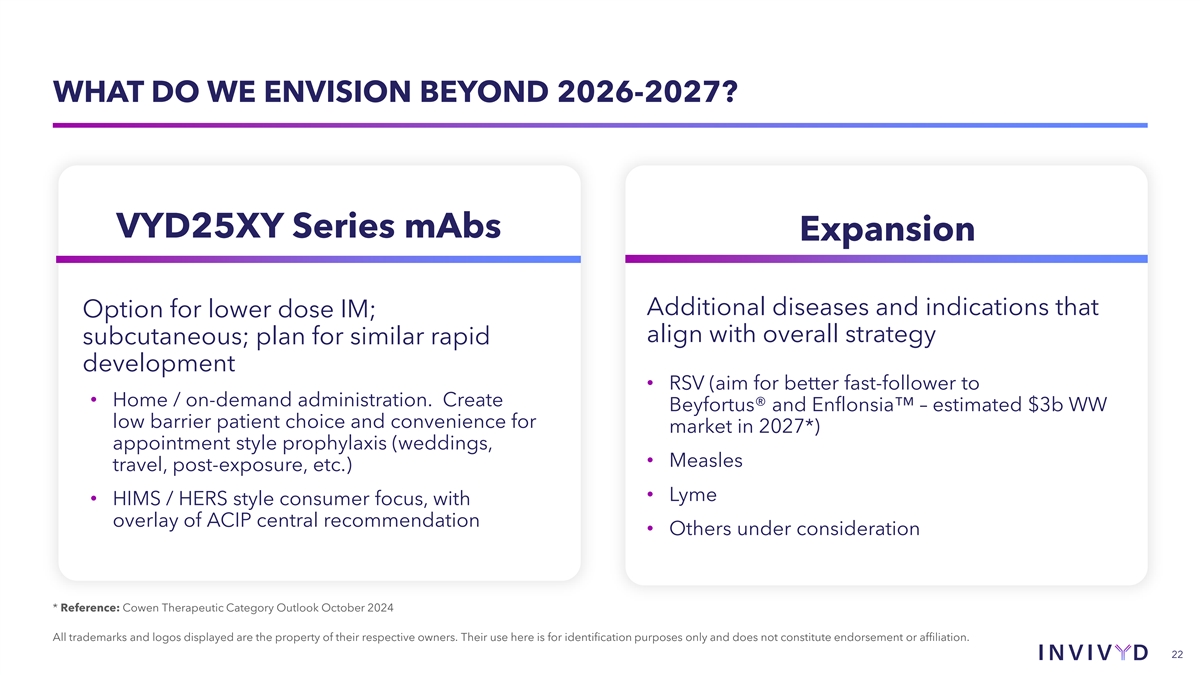
WHAT DO WE ENVISION BEYOND 2026-2027? VYD25XY Series mAbs Expansion Additional diseases and indications that Option for lower dose IM; align with overall strategy subcutaneous; plan for similar rapid consideration development • RSV (aim for better fast-follower to • Home / on-demand administration. Create Beyfortus® and Enflonsia – estimated $3b WW low barrier patient choice and convenience for market in 2027*) appointment style prophylaxis (weddings, • Measles travel, post-exposure, etc.) • Lyme • HIMS / HERS style consumer focus, with overlay of ACIP central recommendation • Others under consideration * Reference: Cowen Therapeutic Category Outlook October 2024 All trademarks and logos displayed are the property of their respective owners. Their use here is for identification purposes only and does not constitute endorsement or affiliation. 22

ENABLE SHIFT IN INFECTIOUS DISEASE PREVENTION TO BEGIN TO REBUILD TRUST IN AMERICAN HEALTHCARE THROUGH CHOICE AND NATURAL IMMUNE SUPPORT Move Away From: Move Toward: • Addressing public health crisis of distrust in • COVID-19 vaccine-only approach to American healthcare prevention • Individual choice on accessing instant • Central / paternal public health posture protection (mandate) • Supra-physiologic protection • Limitation imposed by human immune systems • Natural supplemental immune support through mAbs • mRNA / adjuvanted / pathogen-centric platforms • Massive disruption of the COVID-19 vaccine market 23

FINANCIALS • Q2 2025 PEMGARDA® (pemivibart) net product revenue of $11.8 million • Continued execution of financial discipline and reduction of operating expenses – $26.2 million in Q2 2025 vs. $27.4 million in Q1 2025 and $32.3 million in Q4 2024 • Ended Q2 2025 with approximately $34.9 million in cash and cash equivalents • Entered into $30 million milestone-based loan facility with Silicon Valley Bank (“SVB”) in April 2025 • Goal of near-term profitability (1H 2025) was not met, but remains possible with the upcoming respiratory virus season expected to contribute to topline and planned continued moderation of operating loss • Well-insulated from potential tariffs and most-favored-nation impact on PEMGARDA 24 Source: Invivyd. Data on File.